Martin Kröger, Clarisse Luap, Patrick Ilg
Preprint DOI: 10.20944/preprints202412.1254.v1 (Open Access)
If your browser does not play the movies, download them using a right-click (save link as ..).
| Movie | κ (kappa) | realization | tw | duration | content |
|---|---|---|---|---|---|
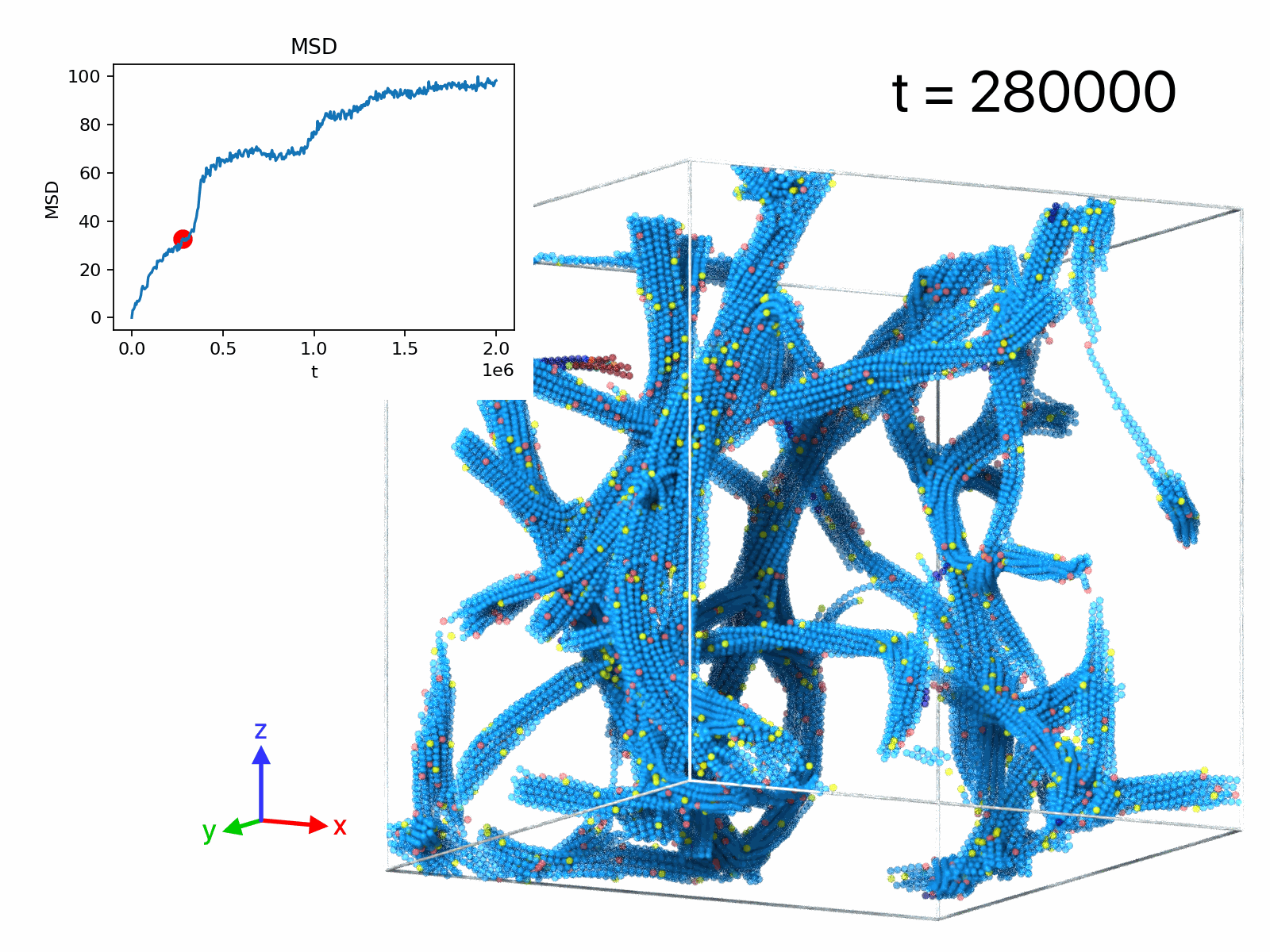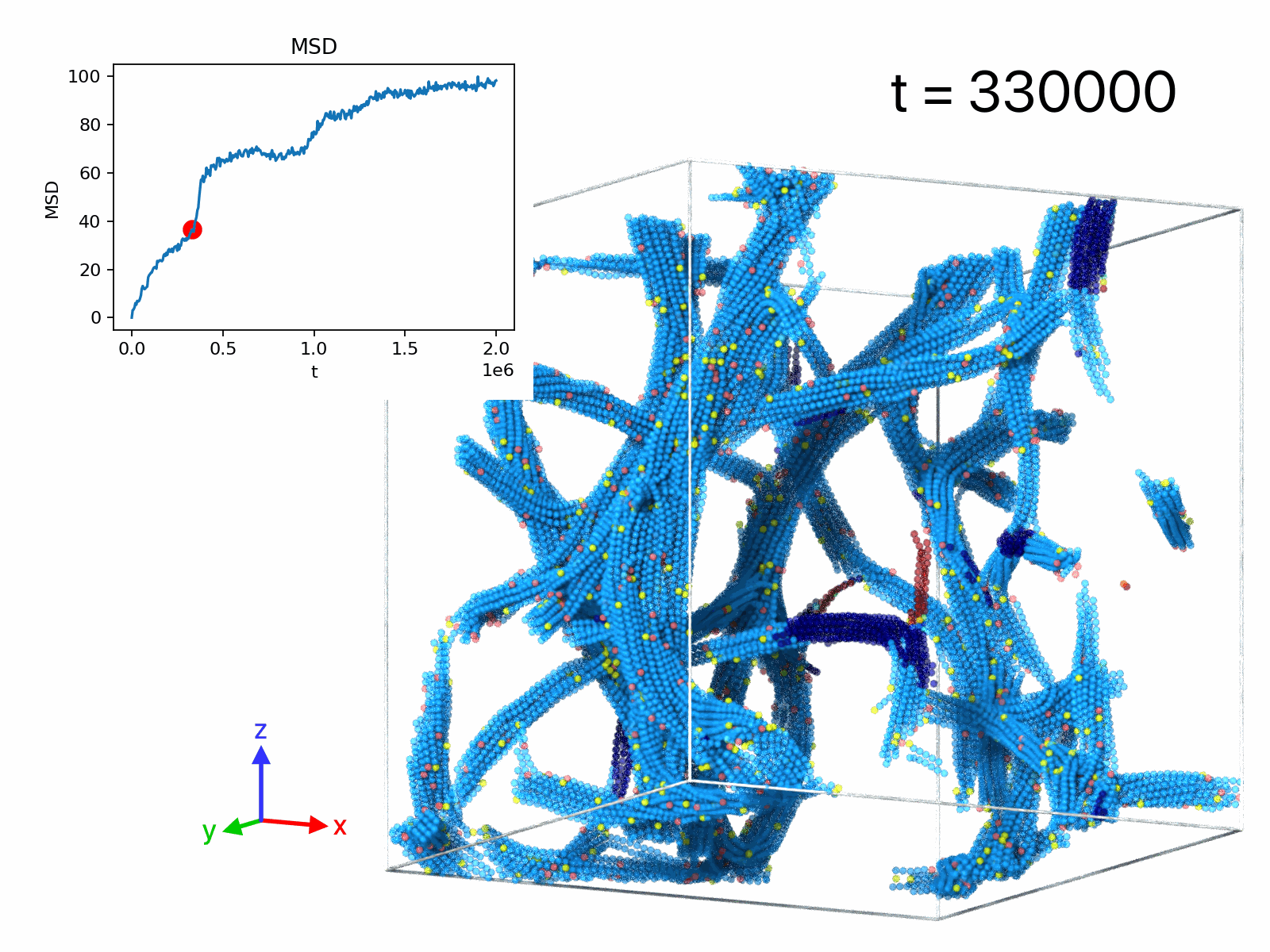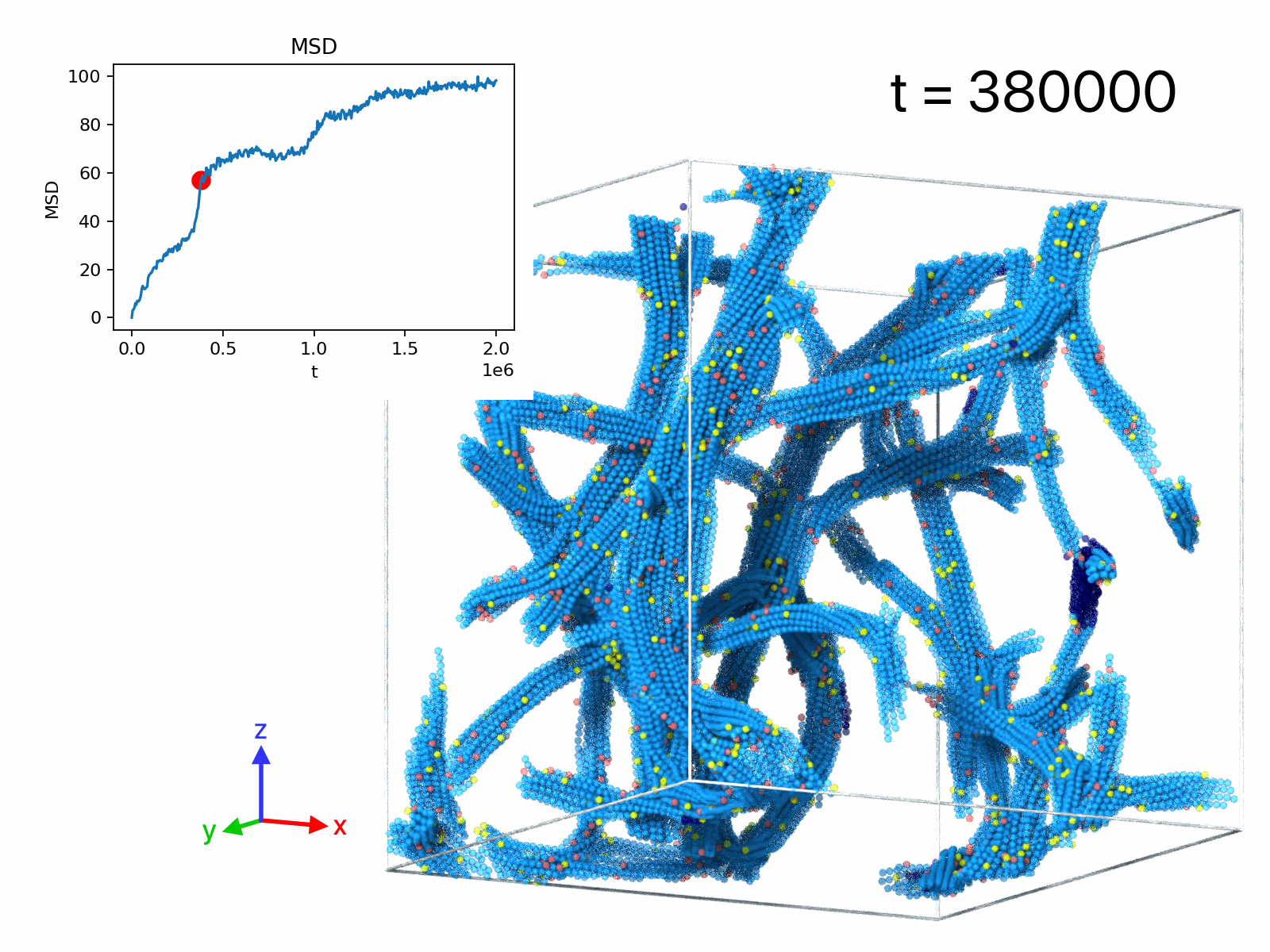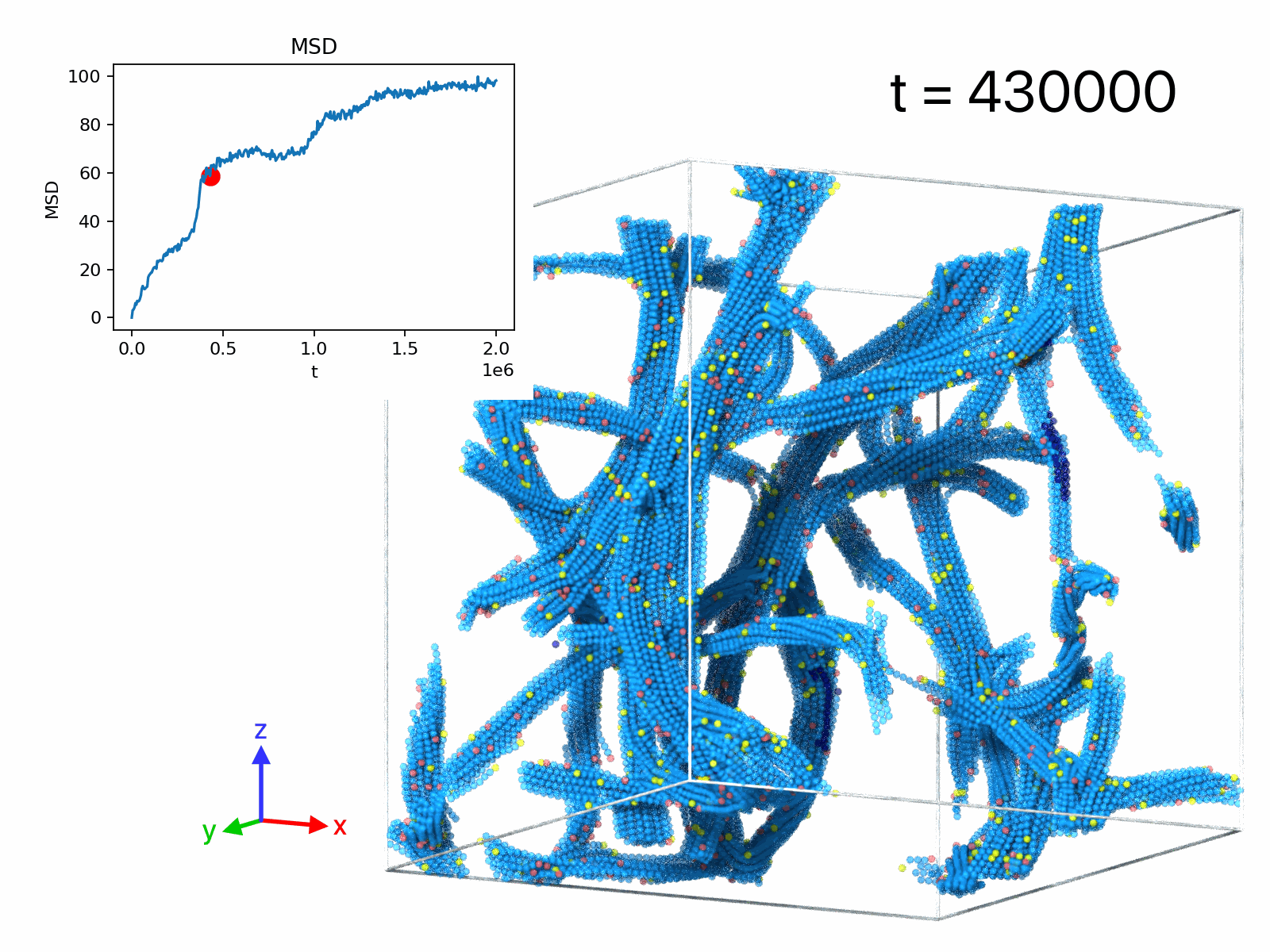
| movie A | 50 | 1 | 1.28× 106 | 1.5× 105 | filament rupture event |
| movie B | 50 | 1 | 105 | 2× 106 | displacements, δt=5× 10^3 |
| movie C | 50 | 1 | 105 | 2× 106 | displacements, δt=2.5× 104 |
| movie D | 50 | 1 | 105 | 2× 106 | displacements, δt=2.5× 105 |
| movie E | 5 | 11 | -103 | 2× 103 | droplet formation |
| movie F | 10 | 11 | -103 | 2× 103 | network formation |
| movie G | 50 | 11 | -103 | 2× 103 | network formation |
| movie E+ | 5 | 11 | 103 | 2× 104 | coarsening dynamics |
| movie F+ | 10 | 11 | 103 | 2× 104 | coarsening dynamics |
| movie G+ | 50 | 11 | 103 | 2× 104 | coarsening dynamics |
| movie H | 5 | 1 | 0 | 105 | droplets and short cylinders |
| movie I | 5 | 6 | 0 | 5× 104 | percolated cylinder |
| movie J | 20 | 1 | 0 | 5× 104 | coarsening dynamics |
{kind=link}
All movies have been created by C.L. using Ovito.
We fitted the ensemble-averaged time series using the logarithmic and power-law expressions mentioned in the manuscript. Both of them involve four parameters for each quantity. The scripts FENE_CB_functions.m (matlab) and FENE_CB_functions.py (python) offer inline functions that allow to evaluate or plot the various quantities (described within the script) versus waiting time (tw) and/or bending stiffness (kappa). Below are rudimentary examples on how to edit and make use of the scripts.
Using matlab, plot the radius of gyration versus waiting time for 6 different kappa values, using the logarithmic fits:
figure;
tw = 10.^linspace(0,5,200);
for kappa = [10 20 30 50 75 100]; semilogx(tw,Rg_power(kappa,tw),'k.-'); hold on; end
Using python, plot the radius of gyration versus waiting time for 6 different kappa values, using the logarithmic fits:
plt.figure()
tw = np.power(10,np.linspace(0,5,200))
for kappa in [10,20,30,50,75,100]:
plt.semilogx(t,Rg_power(kappa,tw),'k.-')
plt.show()
Using matlab, plot the radius of gyration versus waiting time for 6 different kappa values, using the power-law fits:
figure;
tw = 10.^linspace(0,5,200);
for kappa = [10 20 30 50 75 100]; semilogx(tw,Rg_logarithmic(kappa,tw),'k.-'); hold on; end
Using matlab, plot the radius of gyration versus kappa at three different waiting times using the power-law fits:
figure;
kappa = linspace(10,100,100);
for tw = [1e2 1e4 1e5]; plot(kappa,Rg_power(kappa,t),'k.-'); hold on; end
With the functions at hand, you can calculate time-averaged mean values to rate the effect of a chosen averaging interval on the mean values, or to extrapolate quantities to a later waiting time. As the fits are based on data for tw ∈ [104,106] for κ ∈ {0,10,50,75} and tw ∈ [104,105] for κ ∈ {2,5,15,20,30,40,100} the predictions far outside these regimes must be regarded as crude estimates. For example, extrapolating the number of edges (E) to unity makes sense, while extrapolating them to zero is certainly over-stressing the fit function.
The FENE-CB model (manuscript section 4.1.1) studied in this work is coveniently implemented in LAMMPS via
variable rc equal 1.359 # cutoff distance for nonbonded pairs of beads (corresponds to E_coh = 1.4)
variable kappa equal 10 # bending stiffness
atom_style angle
read_data myfile.data nocoeff
mass * 1.0
pair_style lj/cut ${rc}
pair_modify shift yes
pair_coeff * * 3 1.0 ${rc}
bond_style fene
bond_coeff * 30 1.5 1.0 1.0
special_bonds fene
angle_style cosine
angle_coeff * ${kappa}
timestep 0.005
comm_modify cutoff 4.0
neighbor 0.3 bin
fix NVE all nve
fix LANGEVIN all langevin 1 1 2 123456 zero yes
# run etc ..
where it is assumed that you have an initial configuration myfile.data at hand (if not, contact M.K.). The entries within the Atoms section of the data file are: id mol type x y z. The Bonds section lists the permanent intramolecular bonds. The above can serve as the header of your LAMMPS input file.